近日,深圳大学医学部许兴智教授团队与深圳大学总医院巩鹏教授团队合作,在EMBO Journal期刊发表题为“FOXP1 phosphorylation antagonizes its O-GlcNAcylation in regulating ATR activation in response to replication stress”的研究论文。相关内容揭示了叉头框(forkhead box, FOX)转录因子FOXP1促进复制压力下ATR-CHK1活化的新机制,该功能不依赖于FOXP1的转录调控活性,并且由FOXP1的磷酸化修饰和糖基化修饰交互调控。医学部基础医学院副研究员朱雪霏、联合培养博士生高聪文和副教授彭斌为论文的共同第一作者,许兴智教授、巩鹏教授和朱雪霏副研究员为共同通讯作者。
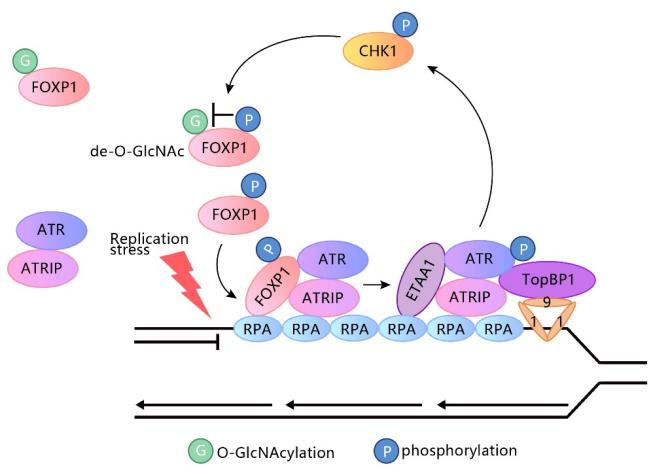
基因组DNA在复制过程中受到多种内源和外源压力的干扰,导致复制叉的减缓或停滞。高效的DNA复制压力应答机制可确保基因组信息的精准复制,ATR信号通路在其中发挥重要作用。ATR作为复制压力感知和应答通路中的关键激酶,通过催化一系列底物蛋白的磷酸化调控多种应答机制以维持基因组的稳定性,包括稳定和重启阻滞复制叉,抑制复制起始,维持dNTP水平以及激活细胞周期检验点等。ATR通路缺陷可以引发细胞功能受损甚至细胞死亡,并在个体水平导致发育异常和肿瘤发生。该项研究鉴定了新的ATR激活调控蛋白FOXP1。FOXP1通过与RPA-ssDNA直接结合募集到阻滞复制叉,并通过蛋白间直接互作促进ATR到染色质的募集以及ATR-CHK1通路的活化。其中FOXP1的O-GlcNAc糖基化修饰抑制FOXP1与ATR的结合,而复制压力下CHK1介导的FOXP1磷酸化修饰拮抗其糖基化修饰,形成ATR-CHK1活化的正反馈调控通路 (工作模式图如下所示)。FOXP1的致病突变体导致ATR-CHK1活化的缺陷以及阻滞复制叉的不稳定,该现象部分解释了FOXP1缺陷导致发育异常和肿瘤发生的致病机制。这是许兴智教授团队继发现先天性小头畸形致病基因编码的ASPM蛋白(PNAS 2022)和多聚(ADP)核糖聚合酶PARP1的UFM1修饰(PNAS 2024)促进ATR-CHK1活化之后的又一新的调控机制。
该研究得到国家自然科学基金和深圳市科技创新委员会基金等项目的资助。
原文链接:https://www.embopress.org/doi/full/10.1038/s44318-024-00323-x
撰稿:朱雪霏 审核:许兴智



 科研动态
科研动态